We have implemented a disease deconstruction pipeline using single cell and spatial technologies applied to inflamed tissues in humans to discover new cell types, cell states and pathways that mediate autoimmune disease. We then use in vitro organoids, other reductionist approaches, and mouse models to define mechanisms and therapeutic insights for the new pathways and cell states. By shifting between humans and mice, our studies unravel the most relevant autoimmune and inflammatory abnormalities in vivo and then interrogate them in experimental systems. Over the past few years, we have identified new pathogenic T cell populations, macrophage states, and stromal cell subsets that drive pathology in autoimmune disease. Recently, we identified a new pathway of complement activation. Below are several of these examples.
We carried out mass cytometry (CyTOF) and single cell RNA sequencing with unbiased clustering of synovial tissue cells in rheumatoid arthritis (RA). Using these approaches, we identified a new population of T helper cells that we have named T peripheral helper (Tph) cells, to distinguish them from T follicular helper (Tfh) cells. Tph cells are found in leukocyte aggregates in chronically inflamed peripheral tissues where they drive extrafollicular B cell differentiation and autoantibody production. Tph cells express high levels of PD1 and ICOS, like Tfh cells, but have low expression of Bcl6 and instead of expressing CXCR5 and localizing in lymph node germinal centers, they express CCR2 and home to peripheral tissues where they secrete CXCL13, the ligand for CXCR5 and IL-21 to provide B cell help and plasma cell differentiation in peripheral autoimmune lesions. Figure below shows Tfh cells (upper) and Tph cells (lower) panels.
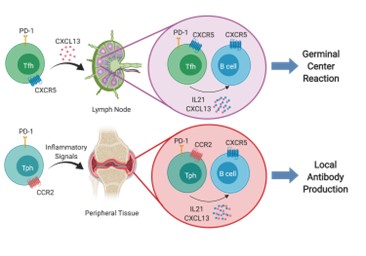
Since our initial studies, Tph cells have been found to be expanded in a range of inflammatory diseases including lupus, autoimmune hepatitis, Celiac disease, juvenile idiopathic arthritis (JIA) and other conditions where autoantibodies are present.
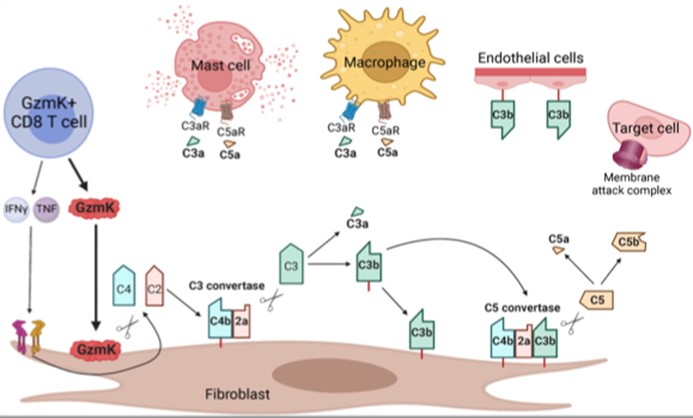
Most previous studies on CD8 T cells have focused on the role of CTL (cytolytic T lymphocytes) expressing granzyme B that cleaves caspases and kills target cells. However, using scRNA-seq of inflamed tissues, we found that the major phenotype of tissue CD8 T cells in humans expressed high levels of granzyme K (GzmK) either without or with only low levels of GzmB. In fact, GzmK CD8 T cells dominate in the inflamed tissues not only in RA, but also in Crohn’s Disease and ulcerative colitis gut, lupus nephritis kidney and other autoimmune diseases. This surprising finding led us to ask what GzmK+ CD8 T cells do. First, we found that rather than CD4+ T cells, these CD8 T cells are the dominant T cell source of IFNγ in RA synovium. More remarkably, we found that GzmK mediates a new pathway of complement activation. GzmK cleaves both complement factor 4 and factor 2 to generate an active C3 convertase (C4b2a) which cleaves factor 3 to yield C3a (the anaphylatoxin) and CD3b (the opsonin) with consequent formation of a C5 convertase and activation of the downstream complement mediated inflammation (C5a and MAC). In inflamed tissues, stromal fibroblasts produce large amounts of C2, C3 and C4 which are the major source of complement produce locally and acted on by GzmK. This represents a tissue focused process that differs from the previously known classical, alternative or lectin pathways of complement activation. In contrast to serum complement derived from the liver, here complement is produced locally in tissues; and further, compared to the proteases that initiate the classical pathway (C1r, C1s), the alternative pathway (CFB) or the lectin pathway (MASP1, MASP2), it is lymphocyte-derived GzmK that drives this new pathway of complement activation.
Macrophages regulate protective immune responses to infectious microbes, but aberrant macrophage activation frequently drives pathological inflammation. To identify regulators of pathological macrophage activation, we analyzed RNA-seq data from human autoimmune disease tissues and identified SLAMF7 as a receptor associated with a super-activated macrophage state in rheumatoid arthritis. This occurs by a two-step process in which IFNγ exposure upregulates SLAMF7 expression following which homotypic engagement of SLAMF7 by SLAMF7 on other cells, drives an exuberant wave of inflammatory cytokine expression exceeding that of macrophages activated by TNF, IFNγ, TLR and other agonists.
Figure (below) shows heat map of gene expression from cultured macrophages after IFNγ stimulation versus SLAMF7 stimulation (expression of multiple chemokines, cytokines, growth factors expressed at the highest levels detected following any type of activation).
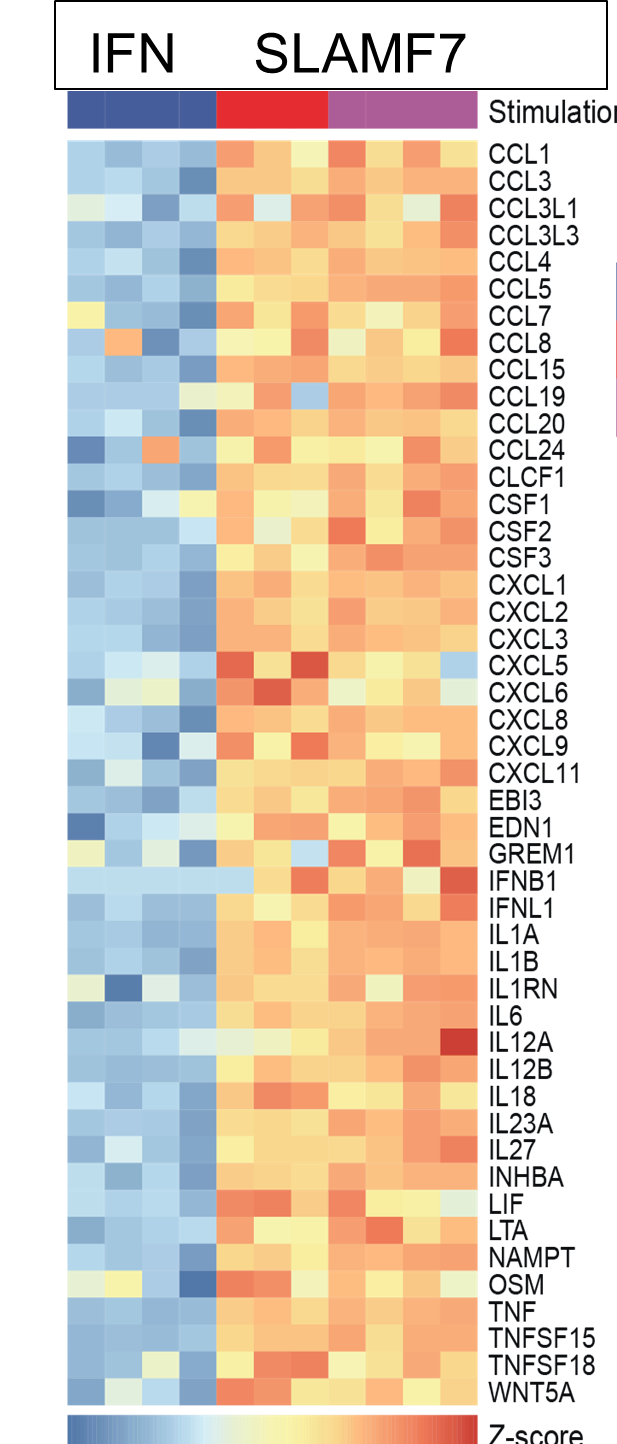
Thus, we termed this a super-activated macrophage state. We observed the SLAMF7-induced super-activated macrophages across a range of diseases including rheumatoid arthritis, Crohn’s disease, and severe COVID-19 pneumonia. This implicates a central role for SLAMF7 in macrophage super-activation across many inflammatory diseases.
While fibroblastic stromal cells were known to play key roles in regulating immune responses in lymph nodes and tumors, our studies are defining their central roles in chronic inflammation in peripheral tissues. Using RA as an example, we defined subsets of inflammatory synovial fibroblasts that are expanded in RA and accounts for over half of all fibroblasts in the disease. These are sublining fibroblasts that express high levels of HLA-DR, IL-6 and many chemokines, all expressed at even higher levels than leukocytes (Mizoguchi et al. Nat. Comm. (2019), Fan et al. Nat. Immunol. (2019). Importantly, we noted that a particular inflammatory subpopulation of fibroblasts in the sublining was oriented around blood vessels. We showed that blood vessel endothelial cell-derived Notch ligands drive Notch signaling on the fibroblasts which imparts their sublining phenotype and that this pathway is essential for inflammatory arthritis. Either deletion or blockade of Notch 3 signaling abrogated inflammatory arthritis in mouse models (Wei et al. Nature (2020).
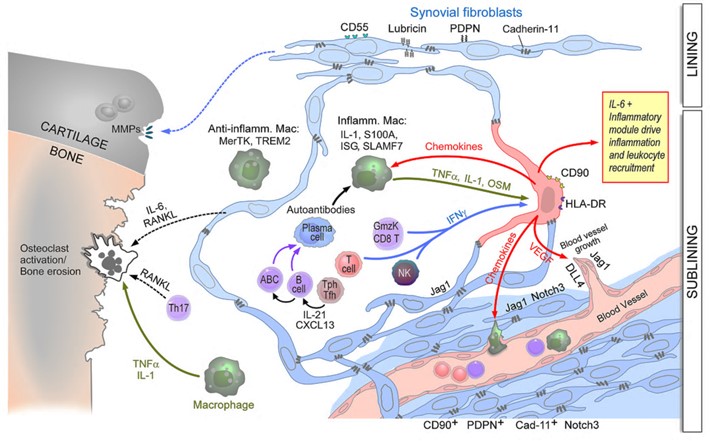
The figure (above) displays the perivascular (sublining) fibroblasts becoming activated by leukocyte-derived cytokines to become the major source of IL-6 and many chemokines and driving degradation of the cartilage, and bone erosion via osteoclast activation.
We then defined the inflammatory fibroblast populations that are shared across multiple tissues and diseases (including RA, IBD, and Sjogren’s Disease) and found that the inflammatory and the Notch signaled fibroblasts are shared across tissues and diseases (Korsunsky et al Med 3: 481 (2022).
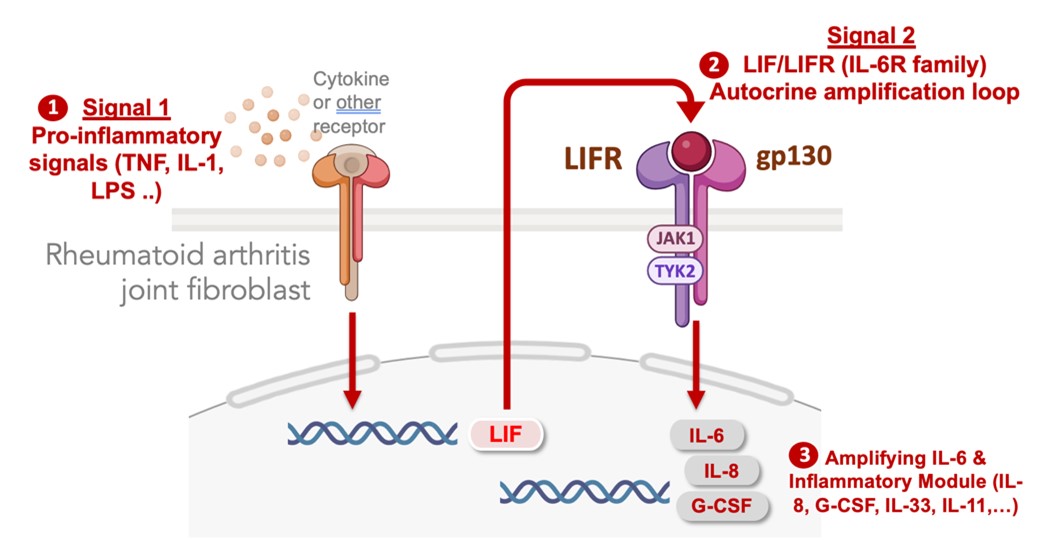
In identifying the pathways that activate stromal cell populations in autoimmune diseases, we defined signal specific gene expression modules in fibroblasts that regulate their secretion of IL-6 in chronically inflamed tissues. IL-6 is part of a larger program of inflammatory factors and transcription factor regulators that are co-expressed as an inflammatory module dependent upon an autocrine feedback loop mediated by cell surface IL-6 receptor family member, leukemia inhibitory factor receptor (LIFR). No matter what exogenous factors activate fibroblasts (e.g. TNF, IL-17, IL-1, LPS) the full activation response is dependent on autocrine induction of LIF which acts on the LIFR to profoundly amplify the activation response. Blocking the LIF-LIFR axis prevents fibroblast activation across a range of stimuli. We are now targeting autocrine amplification pathways in therapeutic approaches to block fibroblast-induced inflammation, fibrosis and tissue damage.
I chaired the Accelerating Medicines Partnership (AMP) RA/SLE consortium for 6 years where we carried out extensive single cell disease deconstruction of RA synovial tissues. We stratified RA patients based on the abundance of cell types in the synovial tissues. Using deep single cell data, we defined receptors-ligand expression, expression of risk loci and using covarying neighbor analysis predicted the interacting cell types. We now lead the successor AMP AIM consortium technology core that is focused on spatial transcriptomics and other single cell analysis now across four diseases, RA, SLE, psoriasis and Sjogren’s Disease. The trove of data generated allows us to continue elucidating the pathogenic interactions driving inflammation and tissue damage in these autoimmune diseases.
Discovery of the αβ T cell receptor (TCR) proteins and genes was one of the most important advances in immunology. During that time, a third rearranging gene, initially thought to be TCRα, but later renamed TCRγ, was identified. It was initially thought to be a vestigial gene as all the rearrangements in αβ T cells were defective. The Brenner lab revealed that an entirely separate lineage of T cells existed that expressed a TCR distinct from TCRαβ. We found that one of the CD3-associated chains on these T cells was the functional protein product of the TCRγ gene, and we found another protein associated in the complex that we named TCRδ. We showed that γδ T cells were a separate population of mature effector T cells that lacked expression of functionally rearranged TCRα or TCRβ genes. We defined many of key functional characteristics of these cells including the fact that they did not recognize peptide-MHC presented antigens. We were the first to propose and identify that their recognition focused on nonpeptide antigens, an idea that has now grown to be a common theme for all 3 groups of innate T cells (γδ T cells, CD1 restricted T cells (below) and MAIT cells). Recently, we found that an invariant subpopulation of γδ T cells that produces IL-17 and expresses PLZF is highly enriched in adipose tissue where it critically regulates Treg cell numbers and adaptive thermogenesis. Mice lacking these γδ T cells cannot maintain body temperature and die in the cold. Example publications below:
The paradigm for T cell recognition was based on the premise that MHC Class I and Class II antigen-presenting molecules present peptides for recognition by αβ TCRs. Thus, it seemed almost impossible when we reported a series of papers in Nature (1) indicating that αβ (and γδ) T cells could recognize foreign antigens in the context of CD1 proteins (which are not encoded in the MHC). Even more remarkable was our finding that the antigens presented were lipids, not proteins. We defined many of the mechanisms that allow the CD1a, b, c and d proteins to intersect with and bind lipid antigens in endosomes. CD1a localizes to and surveys the early endocytic compartment, CD1b and d localize and survey late endosomes and lysosomes, while CD1c promiscuously surveys to bind lipid antigens throughout the endocytic system. We showed how saposins use different mechanisms to load lipids into CD1 proteins. Together, these and related studies outlined the existence of an independent antigen-presentation system for T cells where the universe of lipid rather than peptide antigens are recognized by T cells.
In more recent studies on iNKT cells, we found that self-lipid antigens activate iNKT cells to upregulate FASL which signals IL-1β release from macrophages without inflammasome activation. This important 2-cell pathway enables IL-1β release from infected APCs when microbes can evade inflammasome activation (Donado et al. Cell Reports 2020; 31,107466). Further, we defined a regulatory population of iNKT cells that controls inflammation in adipose tissue by regulating adipose Tregs and M2/M1 macrophage homeostasis and regulates thermogenesis and obesity (Lynch et al 2015) and then found the remarkable ability of adipose iNKT cells to regulate fat burning, weight loss and thermogenesis (Lynch et al 2016). We have defined distinct subsets of iNKT cells in adipose tissue that 1) produce IL-10 as a result of an XBP1s ER stress pathway and 2) produce INFγ which activates NK cells to kill macrophages and control inflammation at steady state (LaMarche et al 2020). Example publications below:
Brigham and Women's Hospital
Hale Building, Room 6002Q
60 Fenwood Road
Boston, MA 02115

