Michael B. Brenner, MD
Elizabeth Fay Brigham Professor of Medicine, Harvard Medical School
Director of the Human Immunology Center at Brigham and Women's Hospital

Alisa Mueller, MD PhD
Postdoctoral Fellow

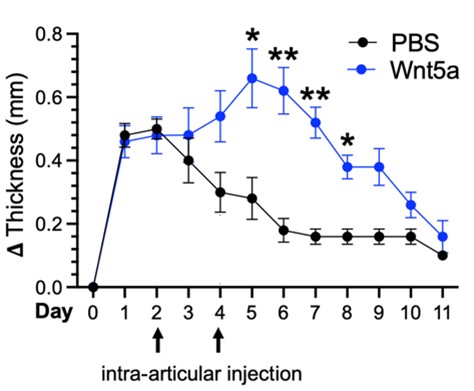
Alisa is an Instructor in Medicine and Associate Physician at Brigham and Women’s Hospital and Harvard Medical School. She received her MD and PhD degrees at Stanford University and completed her medicine residency and rheumatology fellowship at Brigham and Women’s Hospital. In the lab she has explored mechanisms driving stromal inflammation and pathogenicity in rheumatoid arthritis. Her work has identified non-canonical Wnt signaling as a novel mechanism contributing to synovial fibroblast heterogeneity and inflammatory phenotype. In the figure below, non-canonical Wnt pathway activation (blue line) increases the severity and duration of knee swelling in the antigen-induced arthritis mouse model of inflammatory arthritis. Experiments performed in collaboration with Adam Croft’s Laboratory. She has also led a research team to pioneer a CRISPR deletion screen and lentiviral overexpression screen to identify novel regulators of inflammatory fibroblasts.
Heather Faust PhD
Postdoctoral Fellow

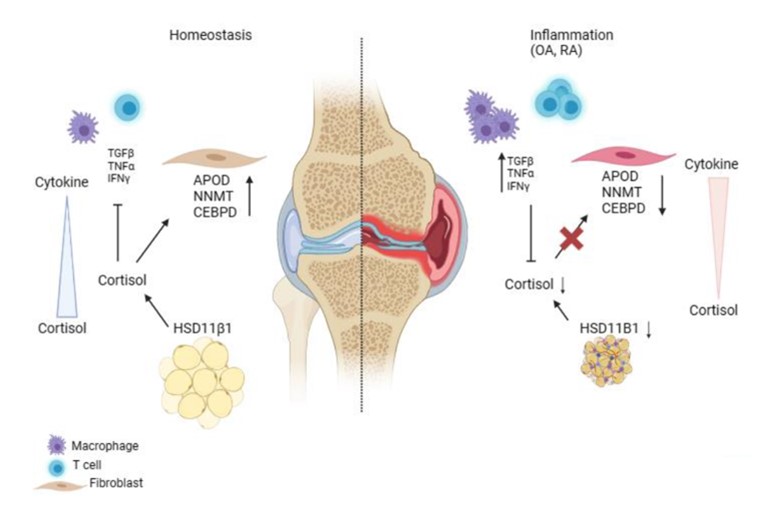
Heather received her PhD from Johns Hopkins School of Medicine in the Cellular and Molecular Medicine program. Her research focused on biomaterials-based approaches to treating osteoarthritis under the mentorship of Jennifer H. Elisseeff. During this time, I became interested in the role of immunology in arthritis and joined Michael's lab for post-doctoral studies. Here, she studies synovial fibroblasts and their pathologic alterations in arthritis. Fibroblasts play critical roles in tissue homeostasis, but in pathologic states they can drive fibrosis, inflammation, and tissue destruction (figure). Little is known about what regulates the homeostatic functions of fibroblasts. We found a gene expression program in healthy human synovial fibroblasts characterized by enhanced fatty acid metabolism and lipid transport. We identified cortisol as the key driver of the healthy fibroblast phenotype and that depletion of adipocytes, which express high levels of Hsd11β1, resulted in loss of the healthy fibroblast phenotype in mouse synovium. Cortisol signaling in fibroblasts mitigates matrix remodeling induced by TNFα- and TGFβ in vitro, while stimulation with these cytokines repressed cortisol signaling and adipogenesis. These findings support the importance of adipocytes and cortisol signaling in driving the healthy synovial fibroblast state that is lost in disease.
Erin Theisen, MD PhD
Postdoctoral Fellow

Erin completed her MD/PhD training at the University of Wisconsin with John-Demian Sauer, where she studied the immunological consequences of Listeria monocytogenes activating the inflammasome. She then completed her dermatology residency at the Harvard Combined Dermatology Residency Program and continues to see patients with autoimmune skin diseases. In the Brenner lab, Erin is working on further defining the Granzyme K-complement pathway, particularly as it relates to skin diseases such as psoriasis and cutaneous lupus, and is developing mouse models to define the in vivo consequences of the GzmK-complement pathway in both skin and arthritis models.
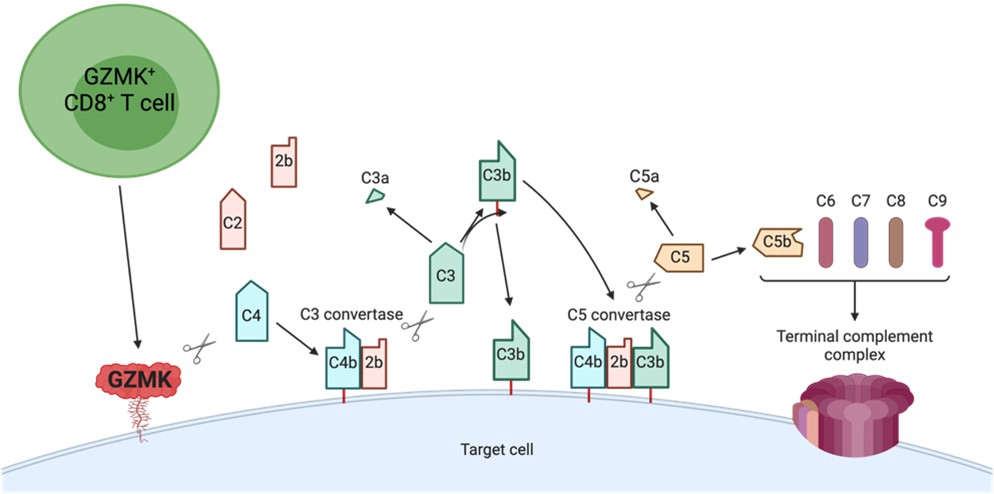
Granzyme K+ (GzmK) T-cells have been identified in numerous autoimmune diseases. GzmK, through direct cleavage of C2 and C4 can form an active C3 convertase on the surface of cells, where it can trigger the main downstream consequences of the complement cascade: opsonization, anaphylatoxin generation, and membrane attack complex formation. Use of GzmK -/- mice in various disease models, including imiquimod dermatitis as a model of psoriasis, and antigen-induced arthritis will further define the contributions of the GzmK-complement pathway to disease pathophysiology.
Hung Nguyen PhD
Postdoctoral Fellow

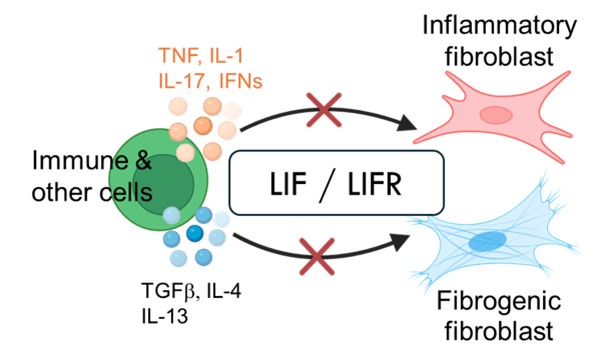
Hung received his PhD in Biology from the Massachusetts Institute of Technology. His postdoctoral work in the Brenner lab, working together with Erika Noss, Edy Kim, and Yunju Jeong, uncovered a key autocrine signaling pathway in pathogenic fibroblasts. This autocrine loop involving leukemia inhibitory factor (LIF) and LIF receptor acts as a master amplifier of fibroblast mediated inflammation and fibrosis.
The autocrine LIF/LIFR acts as a second signal downstream of many pro-inflammatory and pro-fibrotic stimuli in fibroblasts. No matter what the upstream signal is, LIFR signaling is required to drive the pathogenic gene program. Thus, LIFR, a master amplifier of both inflammation and fibrosis, is a promising therapeutic target for chronic and inflammatory diseases, fibrotic disorders and cancer.
Shani T. Gal-Oz, PhD
Postdoctoral Fellow

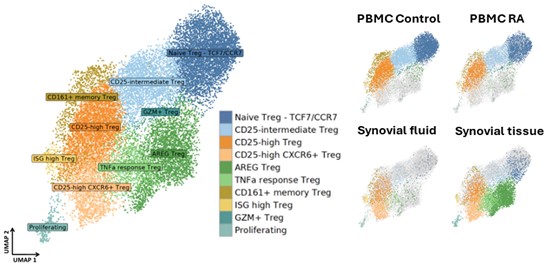
Shani a computational biologist, interested in autoimmune mechanisms sexual dimorphism in health and disease. She completed her PhD at the life sciences department in Ben Gurion University of the Negev in Israel, studying sexual dimorphism in the mice immune system in health and in response to interferon. As a postdoctoral fellow in the Brenner lab and co-mentored by Dr. Ilya Korsunsky, and collaborate closely with Byung-Hee Ko to utilizes single cell RNA sequencing and spatial transcriptomics data to study regulatory T cell sub populations that acts in rheumatoid arthritis during inflammation and their potential interactions with neighboring cells. In the figure below, we identified 10 Treg sub-populations from three rheumatoid arthritis tissues: PBMCs, synovial fluid and synovial tissue; and PBMCs from healthy donors (control) are projected in UMAP embeddings. Two specific sub-populations of interest include CD25 high CXCR6+ Tregs (light orange) that predominantly present in synovial fluid and synovial tissue, and AREG Tregs (green) that almost exclusively appear in the synovial tissue.
Carlos Donado PhD
Postdoctoral Fellow

Carlos obtained his PhD in Immunology at Harvard Medical School in 2021. During my graduate training in Dr. Michael Brenner’s laboratory, I identified an alternative pathway that triggers the release of interleukin-1β (IL-1β), a key orchestrator of anti-microbial immunity whose secretion is typically dependent on activation of inflammasomes. I found that a population of T cells with innate-like functions, invariant natural killer T cells, provide extracellular signals to infected phagocytes to trigger an alternative IL-1β release pathway that supersedes inflammasome activation during infection with inflammasome-evasive microbes.
As a postdoctoral fellow in the Brenner lab, I have focused on elucidating the function of a population of CD8+ T cells defined by high expression of granzyme K, and which makes up the majority of tissue CD8+ T cells in inflamed organs across several inflammatory diseases. In doing so, I discovered a new complement activation pathway that is entirely driven by granzyme K following its cleavage of the complement components C4 and C2 into C4b and C2b. In the figure, GZMK-expressing CD8+ T cells constitutively release GZMK. Secreted GZMK can bind plasma membranes by interacting with heparan sulfate glycosaminoglycans, where it cleaves C4 and C2 generating C4b and C2b. Newly-cleaved C4b molecules can covalently bind membranes through their exposed thioester, associate with C2b, and form membrane-bound C3 convertases. These C3 convertases can cleave C3 into C3a and C3b. Nascent C3b molecules can opsonize target cells or associate with membrane-bound C3 convertases to form C5 convertases that can cleave C5 into C5a and C5b. C5b molecules associate with C6, C7, C8, and C9 to form a terminal complement complex (TCC).
Byung-Hee Koh, PhD
Postdoctoral Fellow

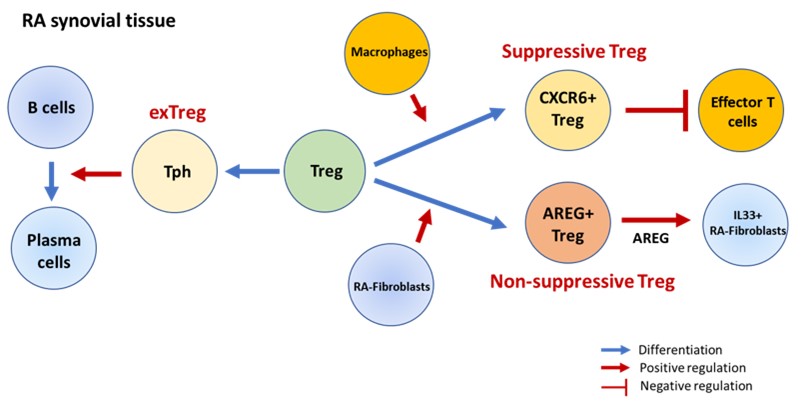
Byung-Hee received his PhD at Indiana University School of Medicine in Dr. Mark Kaplan’s Lab, and currently work as a postdoctoral fellow in the Brenner Lab. His projects focus on determining the heterogeneity and functions of regulatory T cells (Tregs) in patients with rheumatoid arthritis (RA) in collaboration with Shani Gal-Oz. Using combined single cell RNA sequencing analysis and in vitro works, we defined functionally distinct Treg subsets: CXCR6+Tregs and Amphiregulin- expressing Tregs (AREG+Tregs), which are derived through interaction with macrophages or RA-fibroblasts, respectively. Moreover, we also found the conversion of Tregs into peripheral helper T cells (Tph), which induces the differentiation of B cells into plasma cells. Our findings provide a comprehensive characterization of Treg heterogeneity in the RA joint and offer insights into the mechanisms that may control Treg maintenance and function in the context of chronic autoimmune inflammation. In the figure, CXCR6+ Tregs have higher suppressive capability compared to other Treg subsets, while AREG+ Tregs induce IL-33-expressing RA-fibroblasts. Additionally, Tregs can also be converted to Tph cells, acquiring Tph cell function and phenotype while losing those of Tregs.
Ryota Sato, PhD
Postdoctoral Fellow

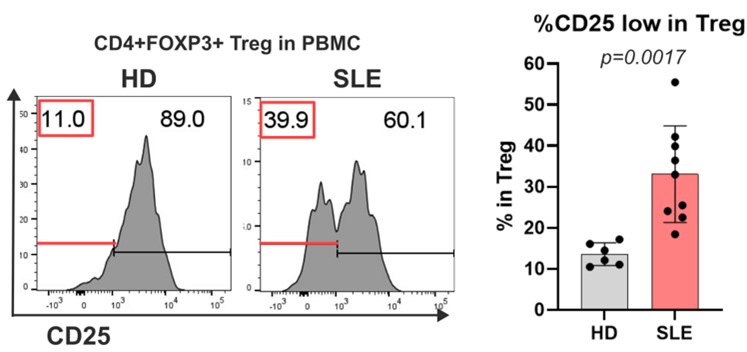
Ryota completed his PhD at Yokohama City University in 2020. He is interested in how our immune system distinguishes self from non-self. His current studies focus on how self-tolerance is dysregulated in various autoimmune diseases, as represented by extraordinary phenotypes of regulatory T cells. We are now defining the pathways that give rise to CD25 low Tregs which account for half of the Tregs in most autoimmune diseases and which are dysfunctional. In the figure, we show CD25 expression on Treg cell in PBMC (peripheral blood mononuclear cell) derived from HD (healthy donor) or SLE (systemic lupus erythematosus) patients highlighting CD25 high and low populations.
Angela Zou
Graduate Student, MD PhD Program, Harvard Medical School

Angela is an MD/PhD student at Harvard Medical School interested in the basis of autoimmunity and inflammation. In the Brenner lab, she is studying the mechanisms governing pathological fibroblast activation in rheumatoid arthritis. Arid5b is a transcriptional co-regulator that associates with demethylases and deacetylaces to regulate gene expression.
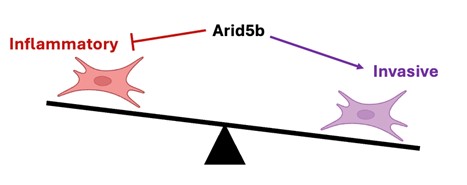
In the figure, we show that fibroblasts can adopt both chronic inflammatory and tissue invasive functions in RA, yet how these behaviors are regulated is not known. We have found that the transcription factor Arid5b inhibits the inflammatory activation of fibroblasts while enhancing their invasiveness, providing insight into how pathologic fibroblast states are differentially programmed.
Suppawat (Kurt) Kongthong
Graduate Student in Immunology, Harvard Medical School

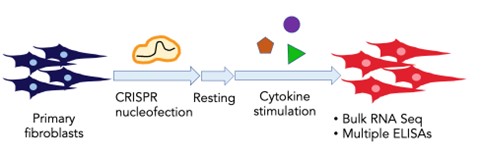
Kurt is a PhD student in the Immunology Program at Harvard Medical School. He uses genetic screens to identify and characterize novel regulators of inflammatory responses in primary fibroblasts. He grew up in Bangkok, Thailand, and received his BA in Biology from Columbia University in New York where he studied the roles of microbial metabolites in gut inflammatory responses with Dr. Nicholas Arpaia and developed methods to engineer and study gut microbes with Dr. Harris Wang. He has now carried out a CRISPR screen in fibroblasts resulting in the identification of novel regulators of pathological fibroblast behavior. In the figure, we show a schematic of the CRISPR screen.
Gerald Watts
Senior Lab Manager

Gerald has an MS degree in Biological Electron Microscopy from the University of Wales, Aberystwyth, and a BS in Biology with Microbiology from Imperial College, London. During his tenure in the Brenner Lab, his research has focused on CD1 lipid antigens in TB and fibroblast biology in rheumatoid arthritis, assisting a large number of researchers, past and present, in successfully bringing their projects to publication. A founding member and technical consultant of the BWH Center for Cellular Profiling core facility, the team provides single cell RNA sequencing and spatial transcriptomics expertise to the MGB community and beyond.
Madison Fairfield
Technician

Madison graduated from Bowdoin College in 2023. She is interested in learning how the immune system works and the various ways to treat it when it malfunctions. She works with Erin Theisen and Heather Faust on a variety of projects involving GzmK, T cells and fibroblasts.

